1. Informasi Umum
Negara Jerman
Jerman merupakan salah satu dari 27 negara anggota Uni Eropa. Oleh karena itu, peraturan perundang-undangan Uni Eropa baik Regulasi, Pedoman maupun Keputusan berlaku di Jerman.
Beberapa area kebijakan yang tidak atau belum diharmonisasi di Uni Eropa dapat diatur secara khusus oleh Jerman.
Peralatan Medis
Berdasarkan Regulation (EU) 2017/745 Peralatan Medis didefinisikan sebagai semua instrumen, peralatan, alat, perangkat lunak, implan, reagen, bahan, atau benda lain yang dimaksudkan oleh produsen untuk digunakan, baik secara sendiri maupun kombinasi, bagi manusia untuk satu atau beberapa tujuan medis tertentu.
Produk berikut ini juga digolongkan sebagai perangkat medis:
- Aksesoris untuk peralatan medis
- Perangkat untuk mengontrol atau mendukung pembuahan;
- Produk yang secara khusus ditujukan untuk membersihkan, mendisinfeksi, atau mensterilkan perangkat
- Produk yang tercantum pada annex XVI
Berdasarkan Regulation (EU) 2017/746 Peralatan medis In Vitro didefinisikan sebagai peralatan medis berupa reagen, produk reagen, kalibrator, bahan kontrol, kit, instrumen, aparatus, peralatan, perangkat lunak atau sistem, baik digunakan sendiri atau dalam kombinasi, yang dimaksudkan oleh produsen untuk digunakan secara in vitro untuk pemeriksaan spesimen, termasuk darah dan sumbangan jaringan, yang berasal dari tubuh manusia, untuk tujuan memberikan informasi mengenai satu atau lebih hal berikut:
(a) mengenai proses atau keadaan fisiologis atau patologis;
(b) mengenai kelainan fisik atau mental bawaan;
(c) mengenai kecenderungan terhadap kondisi medis atau penyakit;
(d) untuk menentukan keamanan dan kesesuaian dengan calon penerima;
(e) untuk memprediksi respons atau reaksi pengobatan;
(f) untuk menentukan atau memantau tindakan terapeutik.
Wadah spesimen juga digolongkan sebagai Peralatan medis diagnostik In Vitro
2. Ketentuan Umum
Medical Device Regulation (MDR)
Regulation (EU) 2017/745 merupakan Regulasi yang mengatur tentang peralatan medis. Peraturan ini bertujuan untuk memperbarui aturan tentang penempatan, ketersediaan dan penggunaan peralatan medis untuk penggunaan manusia dan aksesorisnya di pasar Uni Eropa UE. Peraturan ini mulai berlaku pada 26 Mei 2021. Peralatan medis yang dijual secara online juga harus tunduk pada aturan ini. Beberapa hal yang diatur dalam regulasi ini diantaranya adalah sebagai berikut:
- Persyaratan Keselamatan dan Kinerja Umum
Peralatan medis harus memenuhi persyaratan keselamatan dan kinerja umum yang ditetapkan dalam Annex I untuk meminimalkan risiko terhadap keselamatan dan kesehatan pasien, pengguna, dan pihak ketiga ketika peralatan tersebut dipasok, dipelihara digunakan dengan benar dan digunakan untuk tujuan yang dimaksudkan.
Persyaratan desain dan manufaktur berfokus pada pilihan material, keamanan teknis, biokompatibilitas, sterilitas implan aktif, kemutakhiran, dan lain-lain.
Penujukan kesesuaian dengan persyaratan umum harus mencakup evaluasi klinis sesuai dengan article 61 dari peraturan ini.
- Klasifikasi Resiko Peralatan Medis
Produsen yang ingin menempatkan peralatan medisnya di pasar Eropa harus mengacu pada MDR untuk menentukan kelas risiko yang sesuai untuk produknya. Peraturan ini mengklasifikasikan perangkat menjadi empat kelompok berdasarkan risiko yang mungkin timbul bagi pasien dan pengguna sebagai berikut:
- Kelas I: low risk
- Kelas IIa: medium risk
- Kelas IIb: high risk
- Kelas III: the most critical devices
- Penilaian Kesesuaian
Sebelum mengedarkan peralatan medis di pasar UE, produsen harus melakukan prosedur penilaian terhadap kesesuaian terhadap peralatan tersebut. Prosedur penilaian kesesuaian yang berlaku bergantung pada kelas risiko masing-masing peralatan.
Untuk produk dengan kelas risiko menengah atau tinggi (Kelas IIa, IIb dan III, dan sebagian perangkat Kelas I) diperlukan intervensi dari Notified Bodies. Notified Bodies adalah organisasi yang ditunjuk (oleh negara anggota) yang bertugas menilai kesesuaian terhadap persyaratan keselamatan dan kinerja umum ketika diperlukan penilaian kesesuaian oleh pihak ketiga.
Secara garis besar beberapa hal yang harus dilakukan produsen untuk mendapatkan persetujuan penggunaan CE mark dan menjual produknya di UE adalah sebagai berikut:
- Memeriksa aturan klasifikasi untuk menentukan kelas risiko peralatan medis
- Melakukan GAP analysis untuk menentukan semua persyaratan, kewajiban, file teknis, dan prosedur kesesuaian yang berlaku.
- Menyiapkan dokumentasi teknis (technical file) yang tepat
- Menyiapkan sistem manajemen mutu
- penilaian kesesuaian oleh Notified body (jika diperlukan)
- Membuat declaration of conformity
- Mendaftarkan peralatan medis kepada otoritas pasar terkait.
- CE marking
Perangkat yang dianggap memenuhi persyaratan keselamatan dan kinerja umum harus dibubuhi tanda kesesuaian CE mark ketika dipasarkan. Informasi tersebut harus terlihat jelas, dapat dibaca, dan tidak dapat dihapuskan pada perangkat atau kemasan sterilnya dan pada petunjuk penggunaan, serta pada kemasan penjualan apa pun. Apabila diperlukan penilaian kesesuaian oleh pihak ketiga, CE Mark harus disertai dengan nomor identifikasi dari notified body yang bertanggung jawab atas hasil penilaian kesesuaian.
- Persyaratan Pelabelan
Produk peralatan medis wajib mencantumkan Label pada perangkat medis yang berupa informasi tertulis, tercetak, atau grafis pada perangkat medis, kemasan setiap unit atau kemasan beberapa perangkat medis. Produsen harus memastikan bahwa label produknya memuat seluruh elemen berikut untuk menghindari ketidakpatuhan:
- Nama Produk
- Nomor lot atau nomor seri produk
- Semua detail yang diperlukan pengguna untuk mengidentifikasi perangkat, isi kemasannya, dan tujuan penggunaan perangkat
- Detail kontak produsen (misalnya nama dan alamat)
- nama dan alamat perwakilan resmi, untuk produsen yang berbasis di luar UE,
- informasi sesuai Bagian 10.4.5. dari annex I MDR (Jika relevan)
- Unique Device Identification (UDI) carrier sesuai Bagian c annex VII MDR
- Petunjuk kondisi penyimpanan atau penanganan khusus
- Petunjuk yang jelas mengenai batas waktu penggunaan perangkat dengan aman, atau jika hal ini tidak berlaku, dapat digantikan dengan tanggal pembuatan
- indikasi keadaan steril dan metode sterilisasinya, jika produk dipasok dalam kondisi steril.
- Petunjuk apakah produk tersebut untuk sekali pakai (Jika relevan)
- Petunjuk apakah perangkat dibuat khusus atau ditujukan untuk penyelidikan klinis saja
- Peringatan dan tindakan pencegahan lain yang harus diambil yang perlu segera menjadi perhatian pengguna
- Registrasi produk dan Perwakilan
Produsen yang memiliki tempat usaha terdaftar di luar UE, harus menunjuk perwakilan resmi yang berbasis di Negara Anggota UE sebelum produk mereka dapat dipasarkan. Perwakilan tersebut tersebut harus mendaftar dalam sistem elektronik operator ekonomi.
Produsen harus membubuhkan Unique Device Identifier (UDI) pada perangkat dan kemasan, yang memungkinkan identifikasi dan ketertelusuran. UDI terdiri dari dua bagian, yaitu device identifier (UDI-DI) dan production identifier (UDI-PI) to identify.
Produsen juga bertanggung jawab untuk mendaftarkan produknya dan memperbaharuinya pada database Eropa (EUDAMED) berdasarkan Nomenklatur Alat Kesehatan Eropa (EMDN)
- Kewajiban Manufaktur
Article 10, Produsen harus memiliki sistem manajemen risiko dan manajemen mutu; melakukan evaluasi klinis; menyusun dokumentasi teknis; dan menerapkan prosedur penilaian kesesuaian. Produsen juga bertanggung jawab atas perangkat mereka setelah dipasarkan. Mereka harus mempunyai sistem yang mampu menanggung tanggung jawab keuangan atas kerugian yang disebabkan oleh perangkat medis yang rusak.
Article 15, Setiap pabrikan harus memiliki perwakilan yang ditunjuk yang bertanggung jawab atas kepatuhan terhadap peraturan.
Article 18, Produsen perangkat implan harus menyediakan kartu implan untuk pasien.
Article 19 - 20, Produsen harus membuat pernyataan kesesuaian dan menerapkan tanda CE pada perangkat medis
Article 11, Produsen di luar EU/EEA harus memiliki kontrak dengan perwakilan resmi di EU/EEA.
In Vitro Medical Device Regulation (IVDR)
Regulation (EU) 2017/746 menetapkan ketentuan mengenai penempatan, penyediaan di pasar atau penggunaan peralatan dan aksesori medis diagnostik in vitro untuk digunakan manusia. Peraturan ini juga berlaku untuk studi kinerja mengenai perangkat dan aksesori medis diagnostik in vitro yang dilakukan di UE. Beberapa hal yang diatur dalam regulasi ini diantaranya adalah sebagai berikut:
- Lingkup
Peraturan ini mencakup peralatan dan aksesori medis diagnostik in vitro untuk keperluan manusia. Namun, perangkat yang diproduksi dan digunakan di institusi layanan kesehatan yang sama dikecualikan dari peraturan ini,
- Klasifikasi Peralatan Medis In Vitro
Peralatan Medis In Vitro diklasifikasikan berdasarkan tujuan dan risiko yang melekat pada produk menjadi 4 klasifikasi, yaitu kelas A, B, C dan D. detail lebih lebih lanjut dapat dilihat pada Annex VIII
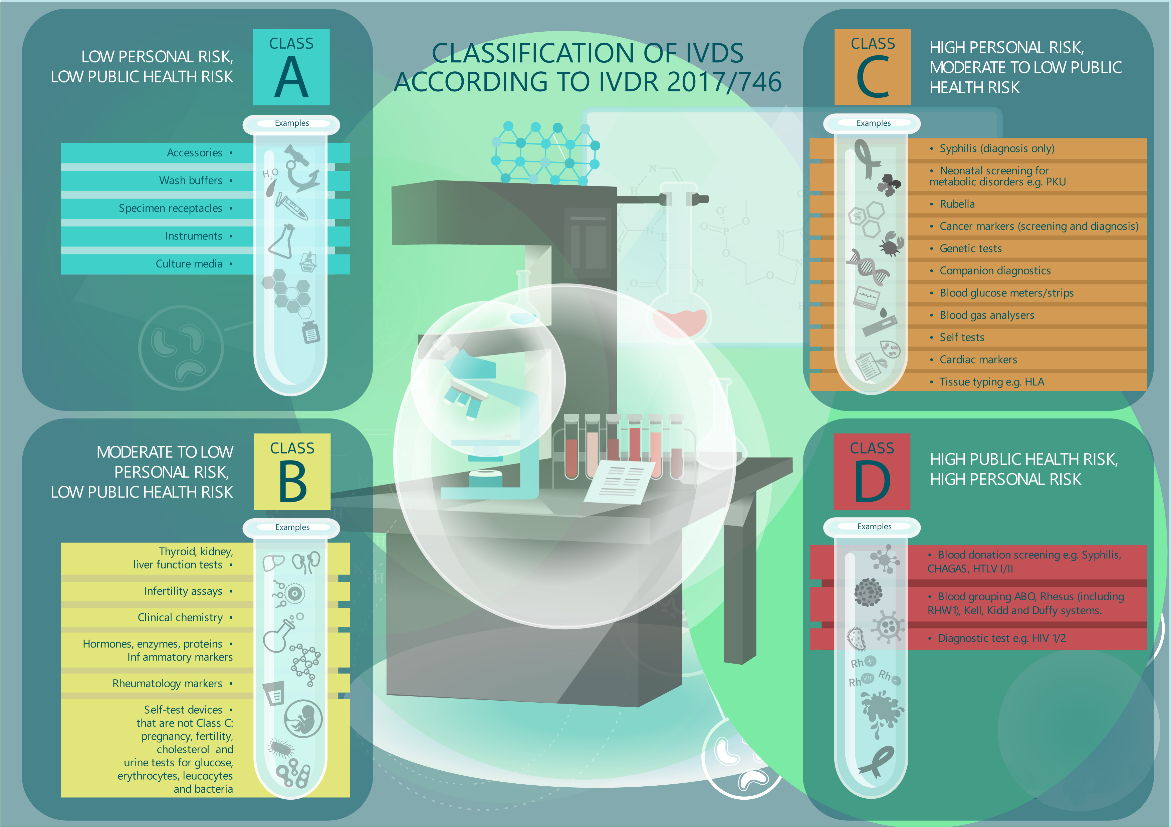
Gambar 1 Infografis Klasifikasi Peralatan Medis Diagnostik In Vitro berdasarkan IVDR 2017/746
- CE marking
Perangkat yang dianggap memenuhi persyaratan keselamatan dan kinerja umum harus dibubuhi tanda kesesuaian CE mark ketika dipasarkan. Informasi tersebut harus terlihat jelas, dapat dibaca, dan tidak dapat dihapuskan pada perangkat atau kemasan sterilnya dan pada petunjuk penggunaan, serta pada kemasan penjualan apa pun. Apabila diperlukan penilaian kesesuaian oleh pihak ketiga, CE Mark harus disertai dengan nomor identifikasi dari notified body yang bertanggung jawab atas hasil penilaian kesesuaian.
- Persyaratan Pelabelan
Produk peralatan medis in vitro wajib mencantumkan Label pada yang berupa informasi tertulis, tercetak, atau grafis pada perangkat medis, kemasan setiap unit atau kemasan. Produsen harus memastikan bahwa label produknya memuat seluruh elemen berikut untuk menghindari ketidakpatuhan:
- Nama Produk
- Nomor lot atau nomor seri produk
- Semua detail yang diperlukan pengguna untuk mengidentifikasi perangkat, isi kemasannya, dan tujuan penggunaan perangkat
- Detail kontak produsen (misalnya nama dan alamat)
- nama dan alamat perwakilan resmi, untuk produsen yang berbasis di luar UE,
- indikasi jumlah isi bersih isi, yang dinyatakan dalam berat atau volume, jumlah numerik, atau kombinasi ketiganya, atau istilah lain yang secara akurat mencerminkan isi kemasan;
- Petunjuk bahwa peralatan ini merupakan alat diagnostic in vitro, atau ‘alat untuk performance study’,
- Unique Device Identification (UDI)
- Petunjuk kondisi penyimpanan atau penanganan khusus
- Petunjuk yang jelas mengenai batas waktu penggunaan perangkat dengan aman, atau jika hal ini tidak berlaku, dapat digantikan dengan tanggal pembuatan
- indikasi keadaan steril dan metode sterilisasinya, jika produk dipasok dalam kondisi steril, atau pernyataan yang menunjukkan kondisi mikroba khusus atau kondisi kebersihan
- Petunjuk apakah produk tersebut untuk sekali pakai (Jika relevan)
- Petunjuk pengoperasian tertentu (Jika relevan)
- Indikasi jika alat ditujukan untuk pengujian mandiri atau pengujian di dekat pasien (Jika relevan)
- Indikasi tegas apabila produk tidak dimaksudkan untuk pengujian mandiri atau pengujian di dekat pasien,
- apabila kit perangkat mencakup reagen individual dan barang yang disediakan sebagai perangkat terpisah, masing-masing perangkat tersebut harus memenuhi persyaratan pelabelan yang tercantum dalam Bagian ini dan persyaratan Peraturan ini;
- Peringatan dan tindakan pencegahan lain yang harus diambil yang perlu segera menjadi perhatian pengguna
- Label untuk peralatan untuk pengujian mandiri harus memuat rincian (i) jenis spesimen yang diperlukan untuk melakukan pengujian (misalnya darah, urin, atau air liur); (ii) bahan tambahan agar pengujian dapat berfungsi dengan baik; (iii) rincian kontak untuk saran dan bantuan lebih lanjut.
- Kewajiban Produsen
- Memantau kualitas, kinerja dan keamanan perangkat
- Menerapkan langkah-langkah yang sesuai dengan tingkat risiko, jenis perangkat dan ukuran perusahaan;
- Memastikan bahwa mereka mempunyai cakupan finansial yang memadai sehubungan dengan potensi tanggung jawab mereka berdasarkan arahan tanggung jawab produkserta manajemen mutu dan sistem pengawasan pasca-pasar.
- Jika terjadi kerusakan karena perangkat yang cacat, perwakilan resmi dari produsen harus bertanggung jawab.
- Registrasi produk dan Perwakilan
Produsen yang memiliki tempat usaha terdaftar di luar UE, harus menunjuk perwakilan resmi yang berbasis di Negara Anggota UE sebelum produk mereka dapat dipasarkan. Perwakilan tersebut tersebut harus mendaftar dalam sistem elektronik untuk Operator Ekonomi.
Produsen harus membubuhkan Unique Device Identifier (UDI) pada perangkat dan kemasan, yang memungkinkan identifikasi dan ketertelusuran. UDI terdiri dari dua bagian, yaitu device identifier (UDI-DI) dan production identifier (UDI-PI) to identify.
Produsen juga bertanggung jawab untuk mendaftarkan produknya dan memperbaharuinya pada database Eropa (EUDAMED) berdasarkan Nomenklatur Alat Kesehatan Eropa (EMDN
Machinery Directive
Directive 2006/42/EC Untuk peralatan medis yang berupa mesin, diharuskan juga memenuhi persyaratan kesehatan dan keselamatan penting sesuai Annex I Directive ini, sejauh persyaratan tersebut lebih spesifik daripada persyaratan persyaratan keselamatan dan kinerja umum yang diatur pada Annex I dari Regulation (EU) 2017/745.
sMedizinprodukte-EU-Anpassungsgesetz / The EU Medical Devices Adaptation Act
merupakan undang-undang yang bertujuan untuk mengadaptasi Regulation (EU) 2017/745 on medical devices dan Regulation (EU) 2017/746 on in vitro diagnostic medical devices kedalam peraturan perundang-undangan alat kesehatan nasional.
Gesetz zur Durchführung unionsrechtlicher Vorschriften betreffend Medizinprodukte / Medical Device Law Implementation Act (MPDG)
adalah undang-undang yang dikeluarkan pada 28 April 2020 untuk melaksanakan dan melengkapi Regulation (EU) 2017/745 MPDG mencakup berbagai aspek, termasuk:
- Tujuan dan lingkup penerapan undang-undang2.
- Kewajiban pelaporan, penempatan di pasar dan pengoperasian produk, serta penyediaan produk di pasar2.
- Persyaratan untuk memulai, perubahan signifikan, dan tindakan korektif2.
- Pengakuan dan pengawasan laboratorium pengujian dan badan penilaian kesesuaian untuk negara ketiga2.
- Persyaratan tambahan untuk uji klinis, studi kinerja, dan uji klinis lainnya2.
- MPDG juga mencakup aturan tentang penyimpanan dokumen dalam kasus penghentian aktivitas bisnis, klasifikasi produk, penentuan status hukum, klasifikasi produk kelas I, dan kewajiban persetujuan untuk uji klinis atau studi kinerja2.
3. Regulasi
Commission Implementing Decision (EU) 2021/1182
Commission Implementing Decision (EU) 2021/1182 adalah keputusan yang dikeluarkan oleh Komisi Eropa pada tanggal 16 Juli 2021 tentang standar harmonisasi untuk peralatan medis yang disusun untuk mendukung Peraturan (EU) 2017/745 dari Parlemen Eropa dan Dewan. Keputusan ini menetapkan standar-standar yang harus dipenuhi olehperalatan medis agar dianggap sesuai dengan persyaratan Peraturan (EU) 2017/746.
Commission Implementing Decision (EU) 2021/1195
Commission Implementing Decision (EU) 2021/1195 adalah keputusan yang dikeluarkan oleh Komisi Eropa pada tanggal 19 Juli 2021 tentang standar harmonisasi untuk peralatan diagnostik in vitro yang disusun untuk mendukung Peraturan (EU) 2017/746 dari Parlemen Eropa dan Dewan. Keputusan ini menetapkan standar-standar yang harus dipenuhi oleh alat kesehatan diagnostik in vitro agar dianggap sesuai dengan persyaratan Peraturan (EU) 2017/746.
Commission Implementing Regulation (EU) 2022/1107
Commission Implementing Regulation (EU) 2022/1107 menetapkan spesifikasi umum mengenai kinerja perangkat kelas D tertentu untuk perangkat diagnostik in vitro. Produsen harus mengikuti spesifikasi atau menunjukkan bahwa solusi alternatif mereka setidaknya setara dalam hal keamanan dan kinerja perangkat.
Implementing Regulation (EU) 2017/2185
Implementing Regulation (EU) 2017/2185 menetapkan daftar kode dan jenis perangkat yang sesuai untuk tujuan menentukan ruang lingkup penunjukan sebagai badan yang diberitahukan di bidang IVD.
Implementing Regulation (EU) 2021/2078
Menetapkan pengaturan yang diperlukan untuk pengaturan dan pemeliharaan EUDAMED. EUDAMED adalah sistem IT yang dikembangkan oleh Komisi Eropa untuk menerapkan MDR dan IVDR. EUDAMED terdiri dari enam modul, yaitu:
- Pendaftaran aktor: Modul ini adalah modul pertama yang harus diisi oleh semua operator ekonomi yang terlibat dalam rantai pasokan alat kesehatan.
- Pendaftaran UDI/alat: Modul ini digunakan untuk mendaftarkan alat kesehatan dan memberikan identifikasi unik (UDI) untuk setiap alat.
- Badan pemberi notifikasi dan sertifikat: Modul ini digunakan untuk mengawasi badan pemberi notifikasi yang menilai kesesuaian alat kesehatan dengan persyaratan peraturan.
- Evaluasi klinis dan kinerja: Modul ini digunakan untuk menyimpan dan mengakses informasi tentang evaluasi klinis dan kinerja alat kesehatan.
- Pengawasan pasca pasar: Modul ini digunakan untuk melaporkan dan memantau insiden yang terkait dengan alat kesehatan, termasuk tindakan korektif lapangan.
- Penilaian kesesuaian dan konsultasi: Modul ini digunakan untuk mengoordinasikan penilaian kesesuaian dan konsultasi antara negara anggota, badan pemberi notifikasi, dan Komisi Eropa.
Medizinprodukte-Agabeverordnung (MPAV) / Medical Devices Dispensing Ordinance
MPAV adalah peraturan yang mengatur penyaluran perangkat medis di Jerman, MPAV juga menyatakan bahwa in vitro diagnostics yang ditujukan untuk deteksi langsung atau tidak langsung dari patogen yang dapat dilaporkan hanya dapat disalurkan kepada dokter, otoritas kesehatan masyarakat, dan penerima istimewa lainnya.
4. Standar
Beberapa contoh standar Uni Eropa
- EN 285:2006+A2:2009 Sterilization – Steam sterilizers – Large sterilizers
- EN 455-1:2000 Medical gloves for single use – Part 1: Requirements and testing for freedom from holes
- EN 455-2:2009+A2:2013 Medical gloves for single use – Part 2: Requirements and testing for physical properties
- EN 455-3:2006 Medical gloves for single use – Part 3: Requirements and testing for biological evaluation
- EN 455-4:2009 Medical gloves for single use – Part 4: Requirements and testing for shelf life determination
- EN 556-1:2001 Sterilization of medical devices – Requirements for medical devices to be designated “STERILE” – Part 1: Requirements for terminally sterilized medical devices
- EN 556-1:2001/AC:2006
- EN 556-2:2015 Sterilization of medical devices – Requirements for medical devices to be designated ”STERILE” – Part 2: Requirements for aseptically processed medical devices
- EN 794-3:1998+A2:2009 Lung ventilators – Part 3: Particular requirements for emergency and transport ventilators
- EN 1041:2008 Information supplied by the manufacturer of medical devices
- EN 1060-3:1997+A2:2009 Non-invasive sphygmomanometers – Part 3: Supplementary requirements for electro-mechanical blood pressure measuring systems
- EN 1060-4:2004 Non-invasive sphygmomanometers – Part 4: Test procedures to determine the overall system accuracy of automated non-invasive sphygmomanometers
- EN ISO 1135-4:2011 Transfusion equipment for medical use – Part 4: Transfusion sets for single use (ISO 1135-4:2010)
- EN 1282-2:2005+A1:2009 Tracheostomy tubes – Part 2: Paediatric tubes (ISO 5366-3:2001, modified)
- EN 1422:1997+A1:2009 Sterilizers for medical purposes – Ethylene oxide sterilizers – Requirements and test methods
- EN 1618:1997 Catheters other than intravascular catheters – Test methods for common properties
- EN 1639:2009 Dentistry – Medical devices for dentistry – Instruments
- EN 1640:2009 Dentistry – Medical devices for dentistry – Equipment
- EN 1641:2009 Dentistry – Medical devices for dentistry – Materials
- EN 1642:2011 Dentistry – Medical devices for dentistry – Dental implants
- EN 1707:1996 Conical fittings with a 6 % (Luer) taper for syringes, needles and certain other medical equipment – Lock fittings
- EN 1782:1998+A1:2009 Tracheal tubes and connectors
- EN 1820:2005+A1:2009 Anaesthetic reservoir bags (ISO 5362:2000, modified)
- EN 1865-1:2010+A1:2015 Patient handling equipment used in road ambulances – Part 1: General stretcher systems and patient handling equipment
- EN 1865-2:2010+A1:2015 Patient handling equipment used in road ambulances – Part 2: Power assisted stretcher
- EN 1865-3:2012 Patient handling equipment used in road ambulances – Part 3: Heavy duty stretcher
- EN 1865-4:2012 Patient handling equipment used in road ambulances – Part 4: Foldable patient transfer chair
- EN 1865-5:2012 Patient handling equipment used in road ambulances – Part 5: Stretcher support
- EN 1985:1998 Walking aids – General requirements and test methods
Lihat selengkapnya disini.
Beberapa contoh standar Jerman.
- DIN EN 1659 In vitro diagnostic systems - Culture media for microbiology - Terms and definitions; German version EN 1659:1996 Edition 1997-01
- DIN EN 13532 General requirements for in vitro diagnostic medical devices for self-testing; German version EN 13532:2002.
- DIN EN 13612 Performance evaluation of in vitro diagnostic medical devices; German version EN 13612:2002.
- DIN EN 13975 Sampling procedures used for acceptance testing of in vitro diagnostic medical devices - Statistical aspects; German version EN 13975:2003.
- DIN EN ISO 1135-3 Transfusion equipment for medical use - Part 3: Blood-taking sets for single use (ISO 1135-3:2016); German version EN ISO 1135-3:2017
- DIN EN ISO 6717 In vitro diagnostic medical devices - Single-use containers for the collection of specimens from humans other than blood (ISO 6717:2021); German version EN ISO 6717:2021
Lihat selengkapnya pada Search Results
5. Lembaga Berwenang
Uni Eropa:
Directorate-General for Health and Food Safety (DG SANTE) adalah direktorat jenderal pada European Commission yang bertanggung jawab atas pemantauan dan implementasi kebijakan dan peraturan Uni Eropa terkait keamanan dan kesehatan pangan.
European Medicine Agency (EMA) adalah adalah sebuah agensi dari EU yang bertanggung jawab atas evaluasi ilmiah, pengawasan, dan pemantauan keamanan obat-obatan. EMA memfasilitasi pengembangan dan akses ke obat-obatan untuk negara-negara di dalam Uni Eropa.
Jerman:
Bundesinstitut für Arzneimittel und Medizinprodukte, - BfArM (Federal Institute for Drugs and Medical Devices) adalah lembaga regulasi medis di Jerman. BfArM beroperasi di bawah Kementerian Kesehatan Federal (BMG) dan bertanggung jawab atas investigasi klinis, pengawasan pasca-pasar, dan pelaporan kejadian peralatan medis.
Paul-Ehrlich-Institute, - PEI (Federal Institute for Vaccines and Biomedicines) adalah lembaga badan regulasi medis dan institusi penelitian untuk vaksin dan biomedis. PEI bertanggung jawab atas penelitian, penilaian, dan otorisasi pemasaran biomedis untuk penggunaan manusia dan produk obat hewan imunologis. Tugasnya juga mencakup otorisasi uji klinis dan farmakovigilans, yaitu pencatatan dan evaluasi efek samping potensial.
6. Informasi Lainnya
- Germany Medical Devices Framework - BfArM
- Germany Regulatory Process for Medical Devices
- Guidance - MDCG endorsed documents and other guidance
- Summaries of EU legislation
- Questions and answers on implementation of the medical devices and in vitro diagnostic medical devices Regulations ((EU) 2017/745 and (EU) 2017/746)
- List of Notified Bodies under Regulation (EU) 2017/745 - Medical devices
- Rapid Alert System for non-food consumer products (RAPEX)